Ubiquitin Ligase COP1 Suppresses Neuroinflammation by Degrading c/EBPβ in Microglia
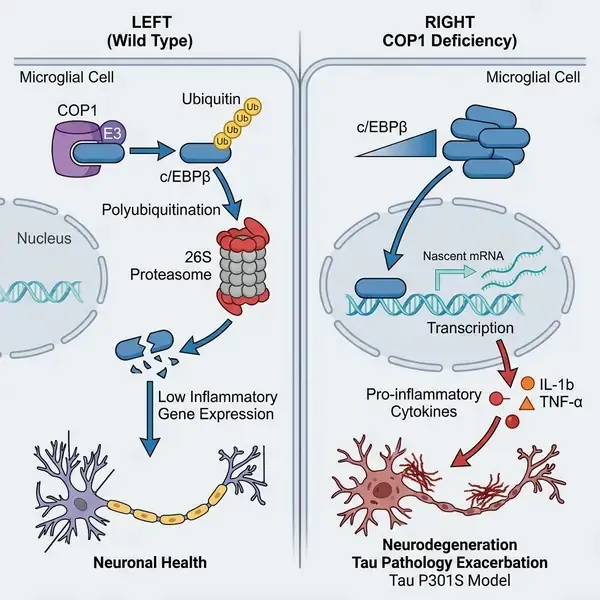
神经炎症主要由小胶质细胞的激活驱动,是包括阿尔茨海默病及相关 Tau 蛋白病在内的神经退行性疾病的核心特征。虽然启动小胶质细胞激活的通路已得到广泛研究,但防止过度或慢性炎症的内源性“刹车”机制仍不十分明确。在发表于《细胞》(Cell) 杂志的一项里程碑式研究中,研究人员确定了 E3 泛素连接酶 COP1(也称为 RFWD2)是小胶质细胞神经炎症的关键负调节因子。该研究表明,COP1 通过靶向转录因子 c/EBPβ 进行蛋白酶体降解,从而抑制促炎基因的表达,维持小胶质细胞的稳态。
研究团队利用遗传小鼠模型和分子生物学技术阐明了 COP1-c/EBPβ 轴。他们发现,COP1 作为 Cullin-RING 泛素连接酶复合物的底物识别组分发挥作用。在生理条件下,COP1 结合炎症信号的主调节因子 c/EBPβ,并促进其泛素化及随后的降解。当小胶质细胞中的 COP1 被遗传缺失时,c/EBPβ 蛋白水平趋于稳定并积累,导致促炎转录程序的强力诱导。这种“高炎症”状态的特征是细胞因子和趋化因子的分泌增加,进而导致神经元损伤。
为了评估这一机制的临床相关性,研究人员采用了 Tau P301S 小鼠模型,该模型模拟了人类 Tau 蛋白病中观察到的病理性蛋白聚集。研究显示,COP1 的缺失显著加剧了疾病表型。与 COP1 野生型对照组相比,缺乏小胶质细胞 COP1 的小鼠表现出加速的神经变性、突触丢失增加以及更明显的认知功能障碍。组织学分析证实,COP1 的缺失导致 Iba1 阳性小胶质细胞激活急剧增加,并伴随神经毒性炎症标志物的激增。这些发现表明,COP1 是防止小胶质细胞转变为慢性激活、有害状态的重要检查点。
这一发现为大脑先天免疫反应的分子调节提供了重要见解。通过确定 COP1-c/EBPβ 通路为核心调节节点,该研究为神经退行性疾病开辟了新的治疗途径。旨在增强 COP1 活性或直接抑制 c/EBPβ 的药理学策略可能有助于减轻神经炎症并减缓阿尔茨海默病等疾病的进展。该研究强调了蛋白质降解途径在维持中枢神经系统免疫平衡中的重要性,并突出了 COP1 作为对抗神经炎症损伤的潜在守护者的作用。