Clustering the protein universe of life using DIAMOND DeepClust
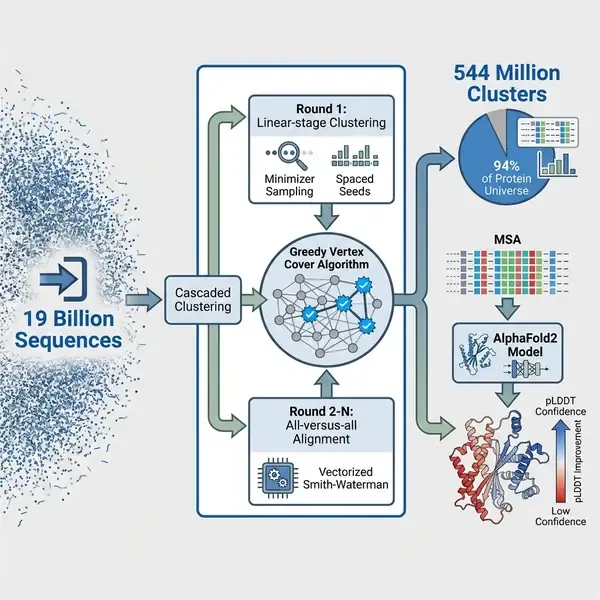
基因组数据的指数级增长已将生物学推向行星级规模,但组织这一庞大“蛋白质宇宙”的计算能力一直相对滞后。在《Nature Methods》发表的一项里程碑式研究中,研究人员推出了 DIAMOND DeepClust,这是一个专为处理数万亿序列设计的级联、超快速聚类框架。通过成功将 190 亿个生物圈蛋白质聚合为 5.44 亿个非单元素簇,该团队不仅展示了前所未有的可扩展性,还为增强人工智能驱动的结构预测提供了关键资源。
行星级基因组学的挑战
随着地球生物基因组计划(Earth BioGenome Project)旨在对 180 万个真核物种进行测序,由此产生的蛋白质空间预计将超过 270 亿条序列。传统的聚类工具(如 CD-HIT、UCLUST 甚至较新的 MMseqs2)面临着根本性的权衡:要么在低序列一致性下牺牲灵敏度,要么由于内存和架构限制而无法扩展到数十亿序列级别。深度聚类(即在低一致性(如 30%)下进行分组以捕捉远程进化关系)尤其耗费资源,通常需要数十万 CPU 小时。
技术创新:级联与并行架构
DIAMOND DeepClust 通过多层策略克服了这些瓶颈。其核心是级联聚类方法,通过多轮比对逐渐提高灵敏度。该过程始于“线性阶段”聚类,利用最小化子采样(minimizer sampling)和多个间隔种子(spaced seeds)快速缩减序列空间。与以往的线性方法不同,DIAMOND DeepClust 采用了优化的双向覆盖标准,确保代表序列和成员序列均满足长度比例要求,这显著提高了保守蛋白质家族的聚类质量。
此外,该软件引入了大规模并行基数排序实现和分布式处理模型。这使得该工具能够跨多个计算节点扩展,绕过了限制以往技术的单节点内存限制。研究人员还实现了一种新的矢量化 Smith-Waterman 算法(基于改进的 SWIPE 方法),将比对推广到多个独立的查询-目标对,在带间隙比对计算中实现了 20% 的性能提升。
基准测试与性能
在使用 NCBI NR 数据库(约 5.46 亿条序列)进行的直接对比中,DIAMOND DeepClust 在深度聚类方面的速度比 MMseqs2 快 36 倍,在单台 64 核服务器上仅需 19 小时即可完成任务。它的速度也比最近开发的 FLSHclust 快 21 倍,同时保持了更高的灵敏度(68.6% 对 49.7%)。当扩展到 190 亿条序列的实验数据集时,该系统利用 27 个计算节点仅用 18 天就完成了聚类,这一壮举此前被认为在计算上是不可逾越的。
映射蛋白质宇宙与增强 AlphaFold2
该研究对 190 亿条序列数据集的分析表明,仅需 5.44 亿个聚类即可捕获蛋白质宇宙的 94%。值得注意的是,研究人员发现了 1.18 亿个新的蛋白质家族,这些家族在目前用于训练结构预测模型的标准数据库 BFD 中并不存在。
为了证明这一扩展数据库的科学价值,团队将其整合到了 AlphaFold2 流程中。对于在现有数据库中代表性不足的序列,使用基于 DIAMOND DeepClust 生成的多序列比对(MSA)显著提高了结构预测的准确性。在 473 个蛋白质的测试集中,平均 pLDDT(预测局部距离差异测试)得分从 52.9 提高到 62.6,凸显了深度聚类在解锁蛋白质组“暗物质”结构见解方面的潜力。
结论
DIAMOND DeepClust 代表了比较基因组学的范式转变。通过提供一个能够扩展到地球生物圈规模的未来型开源解决方案,研究人员为更全面地理解蛋白质进化和功能奠定了基础。随着生命科学迈向万亿序列的地平线,此类高性能工具对于将原始数据转化为生物学发现将是不可或缺的。