Integration of alternative fragmentation techniques into standard LC-MS workflows using a single deep learning model enhances proteome coverage
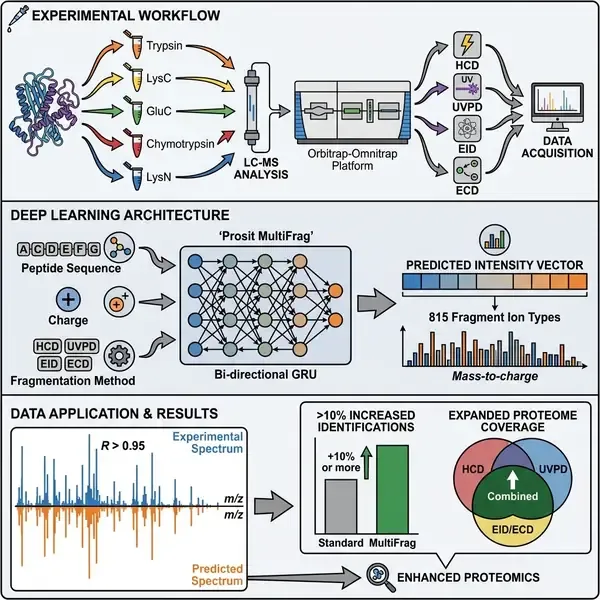
底向蛋白质组学(Bottom-up proteomics)长期以来一直由碰撞诱导解离(CID)技术主导。尽管CID具有极高的速度和重复性,但在表征复杂蛋白质异构体(Proteoforms)和不稳定的翻译后修饰方面存在局限。紫外光解离(UVPD)和电子诱导解离(EID/ECD)等替代碎片化技术能提供互补的序列信息和更丰富的光谱,但由于缺乏自动化仪器和稳健的光谱预测软件,其应用一直受到限制。近日,发表在《Nature Methods》上的一项研究通过开发集成质谱平台和统一的深度学习模型,成功解决了这一难题,实现了对所有主要解离技术碎片强度的准确预测。
研究团队首先通过改进Orbitrap-Omnitrap混合仪器解决了硬件瓶颈。通过引入双阀气体注入系统并优化离子传输电位,他们使Omnitrap能够在液相色谱-质谱(LC-MS)的时间尺度内高效运行。为了构建大规模且多样化的训练数据集,研究人员采用了多酶消化方案(包括Trypsin、LysC、GluC、Chymotrypsin和LysN),并结合离线高pH分级技术。这一过程产生了超过450万张MS2光谱,详尽记录了不同肽段在各种激活方式下的行为。
该研究的核心贡献是开发了改进的Prosit深度学习架构。与以往仅限于碰撞数据的模型不同,这种“MultiFrag”模型利用非结构化输出空间,可预测815种不同碎片离子类型的强度,包括ExD和UVPD特有的自由基物种(a+1, x+1)和非规范碎片(c, z, x)。该模型表现出极高的保真度,在大多数碎片化方法中的中位皮尔逊相关系数均超过0.90。关键在于,该模型具有“碎片化感知”能力,能根据选定的解离方法精准分配离子系列的强度。
通过将该框架集成到FragPipe软件套件的MSBooster模块中,研究人员验证了其科学价值。利用数据驱动的重打分(Rescoring),研究人员观察到肽段和蛋白质鉴定数量显著增加,在数据依赖型(DDA)和数据独立型(DIA)工作流中平均增幅超过10%。在涉及人类、拟南芥和大肠杆菌的DIA实验中,该模型持续提升了鉴定率,其中EID在某些样本中的增幅高达31%。
除了提升鉴定数量,该研究还强调了替代碎片化技术的定性优势。结果表明,UVPD和EID产生的光谱信息更丰富,且受肽段序列特性的影响比CID更小。这项工作通过提供必要的软件支持,有效打破了替代解离技术发展的“僵局”。随着“Prosit_2025_intensity_MultiFrag”模型的公开,蛋白质组学界现在拥有了更强大的工具来表征那些在标准碰撞工作流中难以观察到的复杂蛋白质异构体。